Intuitions behind clone2vec#
The importance of clonal biology#
A clone is a group of cells that all come from a single ancestor cell at some point of development. Stem and progenitor cells divide and give rise to different specialized cell types. By studying clones, we can trace how individual cells contribute to tissues and organs, helping us understand which cells give rise to which structures and how different cell types emerge from the same starting population.
Cells that seem identical at first glance can have very different fates. Even within one tissue, different clones generate a mix of different cell types, revealing hidden functional diversity among cell populations. By analyzing clones in detail, we can better understand development, disease, and how tissues regenerate.
clone2vec: learning clonal contexts from barcoded scRNA-Seq data#
When analyzing barcoded single-cell transcriptomics data, you notice many clones that have the same combinations of cell types or occupy similar transcriptional niches.
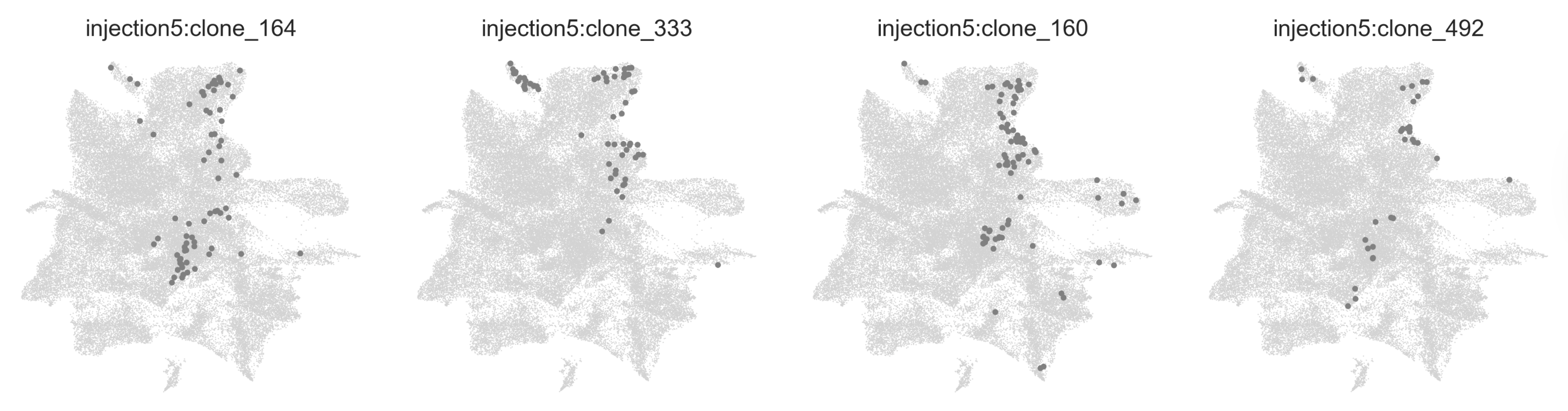
This happens because a cell’s lineage history has a big influence on its gene expression state (even within a single cell type), and these similar-behaving clones are likely to arise from similar (and intuitively, we find, spatially nearby) progenitor cells. We were interested in categorizing these clone types in an unbiased way, so we developed clone2vec, which learns the “clonal context” of cells directly from transcriptional neighborhoods, without using cell type annotations.
Clones similar to each other
Our method is inspired by word2vec, a machine learning model used in language processing. In word2vec, each word is represented as a point in a multi-dimensional space based on its context. Words that appear together in similar contexts have similar meanings — “rat” and “mouse” end up close to each other, just like “apple” and “banana” might.
clone2vec works the same way, but instead of words we use clonal identities of cells. For every cell with a barcode we look at its k nearest neighbors in expression space and record their clonal identities. The model is then trained to predict those neighbor identities from the central cell’s identity. Just like word2vec learns relationships between words based on the company they keep, clone2vec learns relationships between clones based on the neighborhoods their cells occupy in gene expression space. If two clones consistently appear in similar transcriptional neighborhoods, the model places them close together in the clonal embedding space.
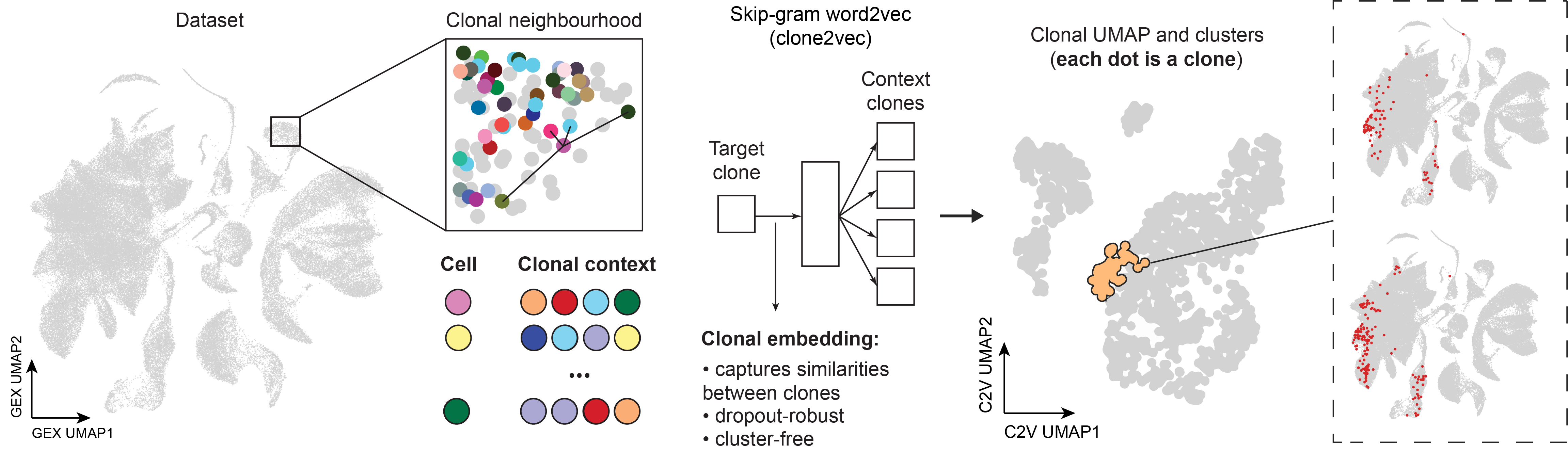
By mapping clones in this way, we can uncover relationships between cell types, predict how clones are functionally related, and explore how different cell lineages contribute to development. Our interactive application clones2cells makes this analysis interactive, allowing users to explore these complex patterns with simple visualizations.
What the clonal embedding actually represents#
The geometry that clone2vec produces is not arbitrary — it has a precise interpretation. At convergence, distances in the embedding space track differences in the pointwise mutual information (PMI) between clones over the cell-cell neighborhood graph. PMI is a standard measure of how much more often two events co-occur than independence would predict: positive PMI means two clones are enriched in each other’s transcriptional company, negative PMI means they avoid each other, and zero means they mix at chance.
Two things follow from this. The embedding cares about how often clones co-occur, not which specific cells co-occurred or how many of them there were. And every clone’s position is fit jointly with every other clone’s, so no clone is ever summarized in isolation. These two properties are what let clone2vec work in the sparse, uneven regime that lineage tracing data always lives in.
Advantages of clone2vec for analyzing cell lineages#
clone2vec embeds clonal data into a continuous vector space, allowing fine-grained analysis of cellular behaviors. Unlike conventional methods, which often rely on binary fate assignments, it indirectly quantifies and compares the proportions of different cell types within clones, capturing subtle variations in cell fate biases. Because it works on transcriptional neighborhoods rather than cell-type labels, it is also robust to the dropout and clustering uncertainty that plague label-based approaches: clones with similar behavior usually share similar transcriptional signatures even within a single cell type, and clone2vec uses that information directly.
Clonal embeddings can be applied to address several critical research questions. Researchers can explore how positional signals influence cell fate decisions by mapping clone behaviors across spatial contexts, leveraging known patterning systems such as the Hox code. Integration of multiple time points of injection allows the identification of specific developmental periods when cell fate decisions become restricted or flexible. And clone2vec aids in elucidating molecular pathways and signaling networks that drive clonal expansion and influence lineage biases, identifying both primary and auxiliary regulatory mechanisms that modulate developmental outcomes.
This scalable framework can be adapted to other tissue systems in embryonic development, immunology, stem cell research, cancer studies, and tissue regeneration.